Oligosaccharides are chains of sugar molecules used in the building of bones, cartilage, skin, tendons and many other tissues in the body. “Oligo” means a few and “saccharide” is a general term for the sugar part of the molecule. In the course of normal life there is a continuous recycling process of building new oligosaccharides and breaking down old ones. The breakdown and recycling process requires a series of special biochemical tools called enzymes.
People with beta-mannosidosis are missing or are low in an enzyme called beta-mannosidase which is essential in breaking down oligosaccharides. When oligosaccharides are not completely broken down they remain stored in the body. The symptoms of beta-mannosidosis are a result of the build-up of oligosaccharides in the tissues in the body. Babies may show little sign of the disease but as more and more cells build up with partially broken down oligosaccharides, symptoms start to appear.
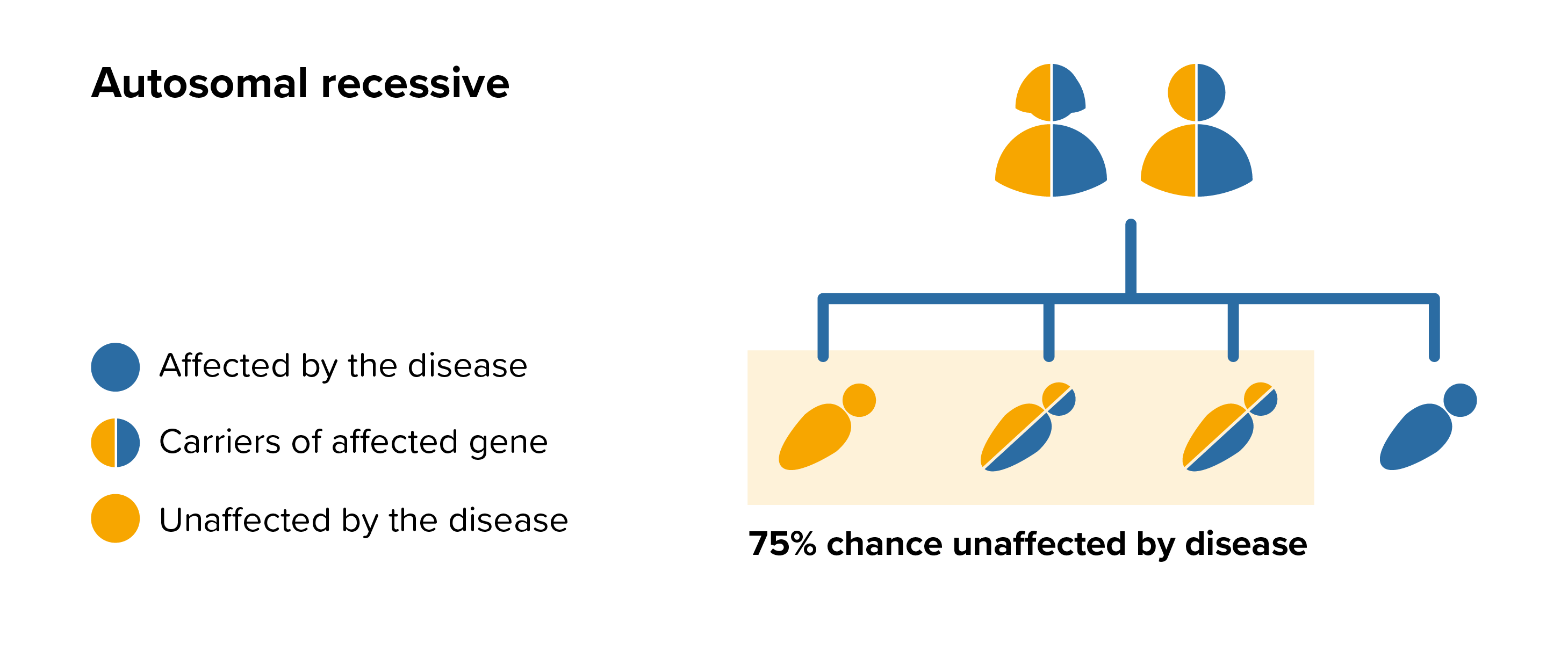 All parents of children with beta-mannosidosis can benefit from genetic counselling, the counsellor can provide advice on the risk to close relatives and to suggest whether the wider family should be informed. To find out during a pregnancy, if the baby is affected by beta-mannosidosis, screening tests can be arranged early on during a pregnancy for those families who already have a child with beta-mannosidosis. Where only one parent is a carrier, they can opt for carrier screening but it is not 100% reliable or accurate and is not possible in all cases.
All parents of children with beta-mannosidosis can benefit from genetic counselling, the counsellor can provide advice on the risk to close relatives and to suggest whether the wider family should be informed. To find out during a pregnancy, if the baby is affected by beta-mannosidosis, screening tests can be arranged early on during a pregnancy for those families who already have a child with beta-mannosidosis. Where only one parent is a carrier, they can opt for carrier screening but it is not 100% reliable or accurate and is not possible in all cases. 




